
Introduction
In late 2021, Singularity underwent a rebranding to Apptainer, changing the command keyword fromsingularity to apptainer, though the majority of subsequent commands and options retained their functionality. Learn more about the changes in the tutorial Apptainer: the container system for secure HPC.
Apptainer is the recommended module for container management, and SCINet users are advised to transition to it for enhanced functionality and support.
What are Containers?
A container can be thought of as a light weight Virtual machine that are in itself a portable linux environment. They help with reproducibility and dependencies required to run a script, program or pipeline.
Initial setup
Before diving into container tasks on HPC infrastructure:
- ensure you checked the available module using the commands:
module avail apptainer module avail singularity - load a module of your choice:
module load apptainer/<version> # e.g., module load apptainer/1.2.5 - and consistently use the appropriate keyword for your commands (in this example: apptainer).
In order to use the container you will need to load the Singularity module on your HPC resource or install it on your local machine. This will vary from HPC resource to HPC resource. You can find what your HPC called the module by using the module avail command. In many cases it will be as simple as the command below.
module load apptainer
On the SCINet HPC, please note that the archival version of Singularity is still accessible.
If required, you can load it using the singularityCE/3.11.4 module.
module load singularityCE/3.11.4
Finding and Downloading a Singularity Container
You can find containers that are compatible with Singularity in several main places:
| resource | description |
|---|---|
| Singularity Hub | Singularity Hub is a dedicated repository for Singularity containers. It hosts a wide range of containers that are specifically built and optimized for use with Singularity. |
| Docker Hub | Docker Hub is a popular repository for Docker images, many of which can be converted to Singularity format using tools like docker2singularity. |
| Biocontainers | Biocontainers is a collection of bioinformatics software packages provided in the form of Docker images. These images can often be converted to Singularity format and used for bioinformatics analysis. |
| Research Groups, Research Projects | Many research groups and projects develop and share their own Singularity containers tailored to their specific needs. These containers may be hosted on their websites, GitHub repositories, or other platforms. |
| Community Forums and Websites | Community forums, mailing lists, and websites related to specific software or fields often provide links to Singularity-compatible containers shared by users and developers. |
The first time you use singularity it will by default put a .singularity folder in your HOME directory which commonly has limited storage space. Therefore it is important that you move that folder to a different location and then create a softlink from your home directory to the new location.
pull from Docker Hub
Docker Hub is a repository of Docker images, hosting a vast collection of containerized software. When searching for a tool on Docker Hub, utilize keywords, filters, and ratings for efficient navigation and selection.
The container used in this example contains the MAKER software tool, which is commonly used for genome annotation.
By pulling and running this Docker container, users can easily utilize the maker tool without needing to install dependencies or manage software versions manually.
Pull the container from Docker hub:
apptainer pull docker://sjackman/maker
Pulling the specified container with apptainer may require some time…
2024/04/29 15:11:46 info unpack layer: sha256:a3ed95caeb02ffe68cdd9fd84406680ae93d633cb16422d00e8a7c22955b46d4 INFO: Creating SIF file...
and upon completion, it will generate a Singularity Image Format (SIF):
ls
maker_latest.sif
To run a containerized tool, use the exec command with the apptainer keyword:
apptainer exec --bind $PWD maker_latest.sif maker --help
This command executes the maker tool inside the maker_latest.sif container while binding the current directory ($PWD) to the container’s filesystem and passing the --help argument to the maker tool.
see the expected output
MAKER version 2.31.6
Usage:
maker [options] {maker_opts} {maker_bopts} {maker_exe}
Description:
MAKER is a program that produces gene annotations in GFF3 format using
evidence such as EST alignments and protein homology. MAKER can be used to
produce gene annotations for new genomes as well as update annotations
from existing genome databases.
The three input arguments are control files that specify how MAKER should
behave. All options for MAKER should be set in the control files, but a
few can also be set on the command line. Command line options provide a
convenient machanism to override commonly altered control file values.
MAKER will automatically search for the control files in the current
working directory if they are not specified on the command line.
Input files listed in the control options files must be in fasta format
unless otherwise specified. Please see MAKER documentation to learn more
about control file configuration. MAKER will automatically try and
locate the user control files in the current working directory if these
arguments are not supplied when initializing MAKER.
It is important to note that MAKER does not try and recalculated data that
it has already calculated. For example, if you run an analysis twice on
the same dataset you will notice that MAKER does not rerun any of the
BLAST analyses, but instead uses the blast analyses stored from the
previous run. To force MAKER to rerun all analyses, use the -f flag.
MAKER also supports parallelization via MPI on computer clusters. Just
launch MAKER via mpiexec (i.e. mpiexec -n 40 maker). MPI support must be
configured during the MAKER installation process for this to work though
Options:
-genome|g {file} Overrides the genome file path in the control files
-RM_off|R Turns all repeat masking options off.
-datastore/ Forcably turn on/off MAKER's two deep directory
nodatastore structure for output. Always on by default.
-old_struct Use the old directory styles (MAKER 2.26 and lower)
-base {string} Set the base name MAKER uses to save output files.
MAKER uses the input genome file name by default.
-tries|t {integer} Run contigs up to the specified number of tries.
-cpus|c {integer} Tells how many cpus to use for BLAST analysis.
Note: this is for BLAST and not for MPI!
-force|f Forces MAKER to delete old files before running again.
This will require all blast analyses to be rerun.
-again|a recaculate all annotations and output files even if no
settings have changed. Does not delete old analyses.
-quiet|q Regular quiet. Only a handlful of status messages.
-qq Even more quiet. There are no status messages.
-dsindex Quickly generate datastore index file. Note that this
will not check if run settings have changed on contigs
-nolock Turn off file locks. May be usful on some file systems,
but can cause race conditions if running in parallel.
-TMP Specify temporary directory to use.
-CTL Generate empty control files in the current directory.
-OPTS Generates just the maker_opts.ctl file.
-BOPTS Generates just the maker_bopts.ctl file.
-EXE Generates just the maker_exe.ctl file.
-MWAS {option} Easy way to control mwas_server for web-based GUI
options: STOP
START
RESTART
-version Prints the MAKER version.
-help|? Prints this usage statement.
pull from Singularity Hub
Singularity Hub has transitioned to DataLad in early 2024 involves a significant change in website layout. However, the archival containers from Singularity Hub are still accessible for those who need them.
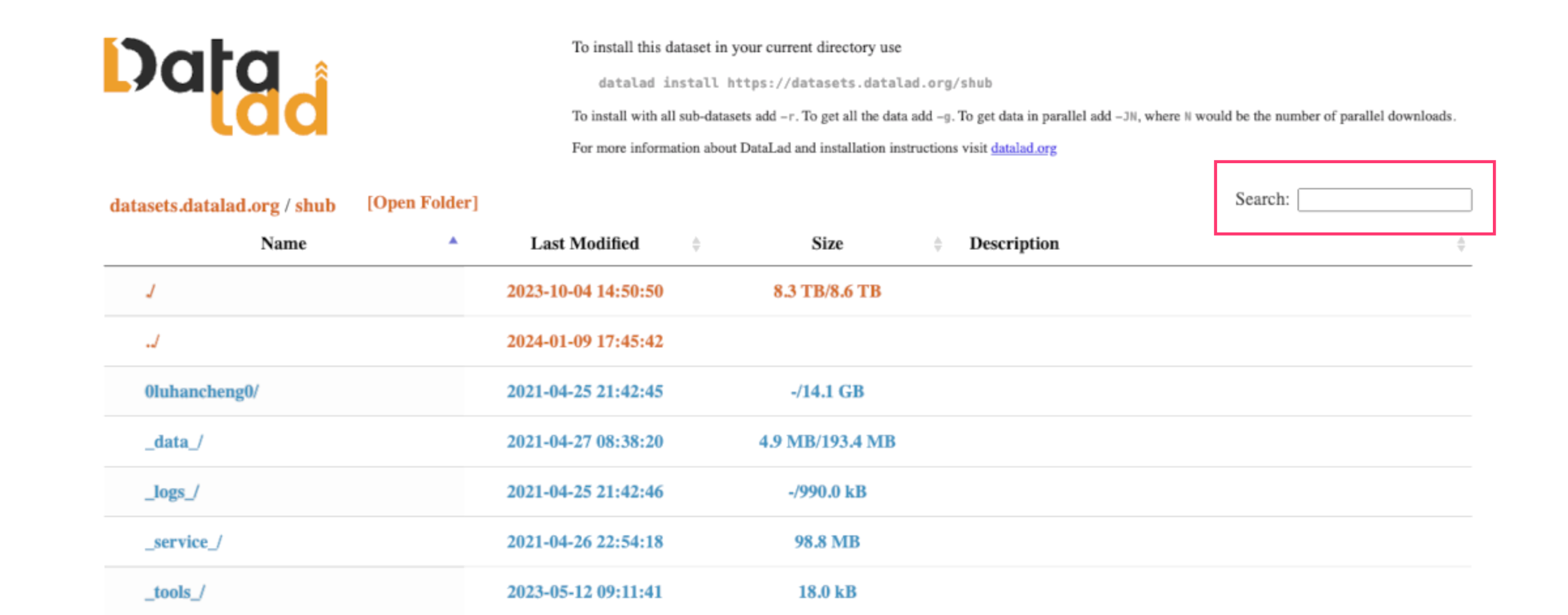
The new DataLad interface presents a simpler directory structure, displaying collections of datasets in a table format.
- You can find collections under clearly labeled folders, such as “data,” “logs,” and others, with metadata like size and last modified date.
- The search bar on the DataLad website facilitates locating specific datasets or tools.
- Additionally, instructions on how to install datasets from the DataLad server are displayed in the top-right corner.
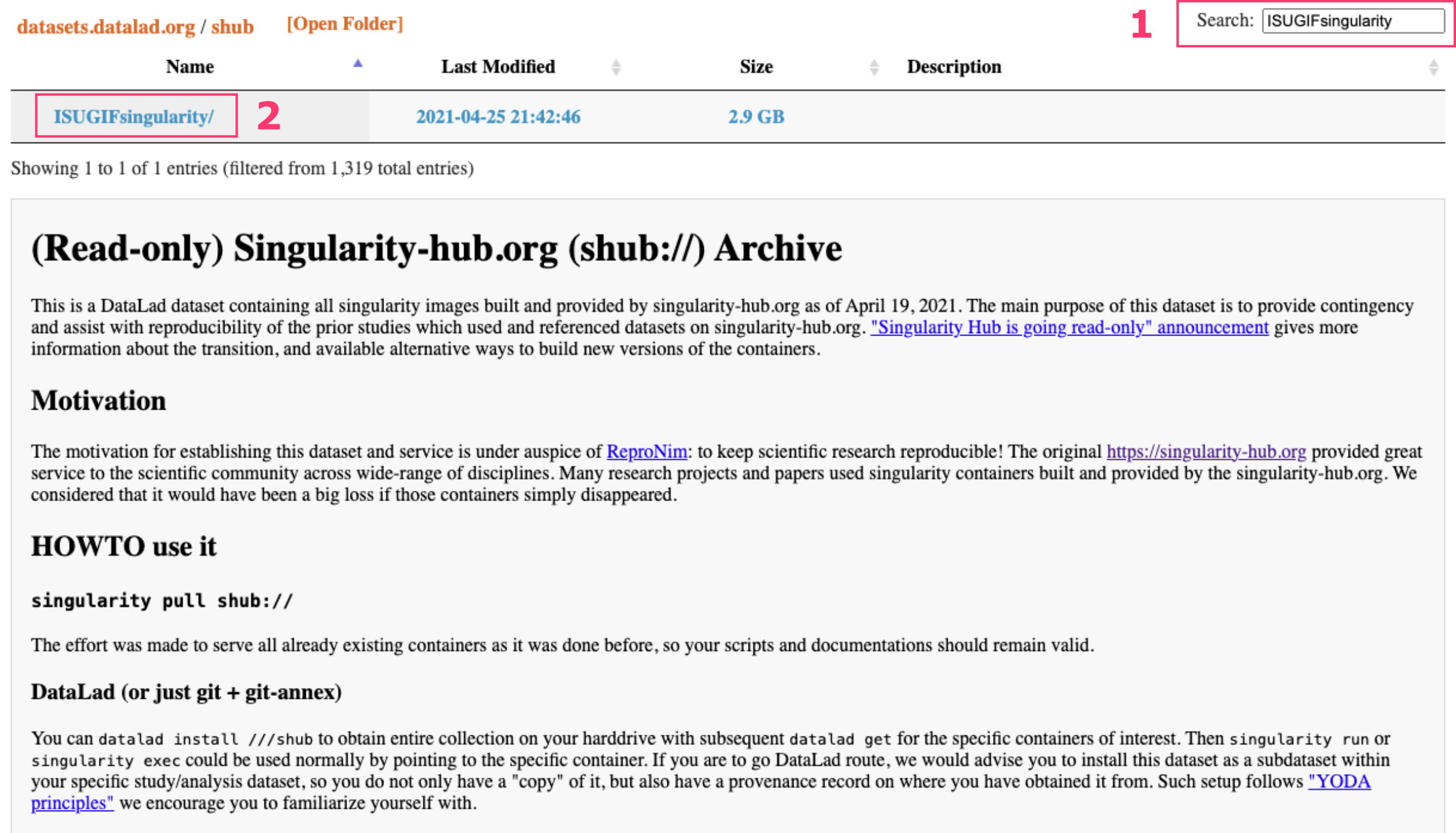
First go to DataLad and locate the container you want through the search box located on the top-right corner of the page.
Archived containers from Singularity Hub are now accessible via shub:// and can be pulled using standard Apptainer commands. To find and pull a specific container, like utilities version 1.0.1, search for the repository name ISUGIFsingularity.
N avigate by clicking through the nested folders to the desired container, and then copy the blue path displayed at the top. This path is the reference you’ll use with shub:// to pull the container.
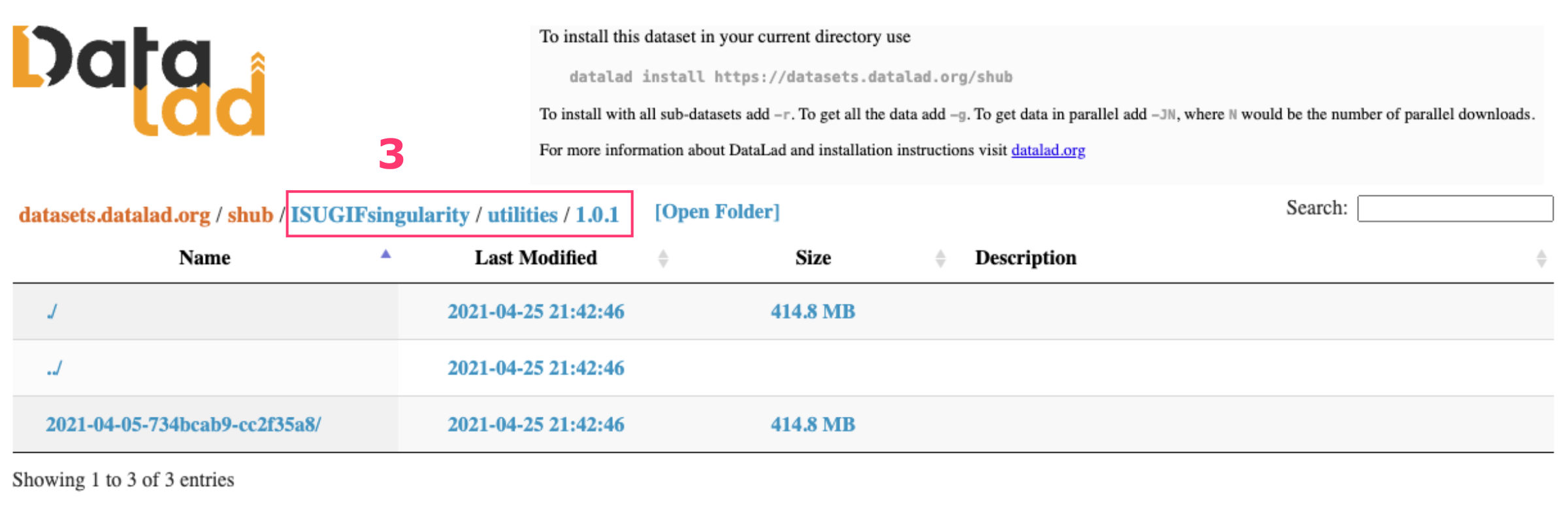
Unfortunately, the copied path (ISUGIFsingularity / utilities / 1.0.1) requires some adjustments:
- you need to replace the last slash
/with a colon:to specify the container version - and remove all white spaces.
The adjusted path will look like ISUGIFsingularity/utilities:1.0.1, ready for your apptainer command:
apptainer pull shub://ISUGIFsingularity/utilities:1.0.1
INFO: Environment variable SINGULARITY_CACHEDIR is set, but APPTAINER_CACHEDIR is preferred INFO: Downloading shub image 392.2MiB / 392.2MiB [=======================================================================================] 100 % 65.4 MiB/s 0s
ls
utilities_1.0.1.sif
If you get a CERTIFICATE_VERIFY_FAILED: error then you can set your python certificate verification to off.
export PYTHONHTTPSVERIFY=0
Direct execution of Singularity containers
Containers often have runscripts that will provide you with useful information on how to use the container. The run scripts get initiated by executing the image as follows:
./ISUGIFsingularity-utilities-master-1.0.1.simg
Which in this case produces a list of files that can be called by the image.
colsum createhist.awk intervalBins.awk nb nbsearch new_Assemblathon.pl readme README.md seqlen.awk summary.sh
As I mentioned above, these are useful scripts often used in our group. One script borrowed from the Assemblathon paper, new_Assemblathon.pl, we use when evaluating genome assemblies and want to get a summary of the assembly statistics. To use this script via the container we can execute it in the following manner.
singularity exec ISUGIFsingularity-utilities-master-1.0.1.simg new_Assemblathon.pl Spirochaete.fasta
This way of executing containers is kind of tedious as it requires so much more than just the new_Assemblathon.pl script that you could just download and place it somewhere in your path. Later in this tutorial, we will show you how to create a bash script wrapper that will simplify calling the function from within the container. In most cases creating a container for a simple script (perl, python, bash) doesn’t make sense as it creates a small overhead to load singularity and the container before executing the script. Where containers shine, is when your script or pipeline requires lots and lots of software prerequisites (for example: a specific version of perl, blast, qiime, samtools, etc)
We ran the new_Assemblathon.pl script from the container on a spirochaete genome we had handy and it produced the desired output.
---------------- Information for assembly '/Users/severin/Downloads/Spirochaete.fasta' ----------------
Number of scaffolds 1
Total size of scaffolds 3251735
Longest scaffold 3251735
Shortest scaffold 3251735
Number of scaffolds > 1K nt 1 100.0%
Number of scaffolds > 10K nt 1 100.0%
Number of scaffolds > 100K nt 1 100.0%
Number of scaffolds > 1M nt 1 100.0%
Number of scaffolds > 10M nt 0 0.0%
Mean scaffold size 3251735
Median scaffold size 3251735
N50 scaffold length 3251735
L50 scaffold count 1
scaffold %A 25.62
scaffold %C 24.57
scaffold %G 24.21
scaffold %T 25.60
scaffold %N 0.00
scaffold %non-ACGTN 0.00
Number of scaffold non-ACGTN nt 0
...
This output has been truncated
How to install and use Singularity on your local machine (Mac)
If you want to explore containers on your local Mac you canfFollow the directions on this website http://singularity.lbl.gov/install-mac
Note: You may need to issue the following command if you update your operating system or get a new computer.
Note you may need to allow Oracle permission via your security settings if you are on a mac. See this website
Starting a singularity Virtual Machine (VM) instance on the Mac
If you are using your local machine, this will allow you to not only execute containers but also build containers from recipes and test them out.
mkdir singularity-vm
cd singularity-vm
vagrant destroy
vagrant init singularityware/singularity-2.4
vagrant up
vagrant ssh
Running a command using a Singularity container. (Same as above)
singularity exec ISUGIFsingularity-utilities-master-1.0.1.simg new_Assemblathon.pl Spirochaete.fasta
transferring files off and onto a local vagrant virtual machines
You probably started your vm and realized that you can’t access any of your files to try out the command above.
If you are running a VM locally to use singularity, you can transfer files to and from your VM using scp and the VM private key.
Change into the folder you initiated your vagrant vm singularity-vm and run the vagrant ssh-config command to get the private key
vagrant ssh-config
OUTPUT
Host default
HostName 127.0.0.1
User vagrant
Port 2222
UserKnownHostsFile /dev/null
StrictHostKeyChecking no
PasswordAuthentication no
IdentityFile /Users/severin/singularity-vm/.vagrant/machines/default/virtualbox/private_key
IdentitiesOnly yes
LogLevel FATAL
Then using that key scp files off of the virtual box from a newly opened terminal that is not in the VM.
scp -P 2222 -i /Users/severin/singularity-vm/.vagrant/machines/default/virtualbox/private_key vagrant@127.0.0.1:/home/vagrant/recipe .
or transfer a file to the VM.
scp -P 2222 -i /Users/severin/singularity-vm/.vagrant/machines/default/virtualbox/private_key ~/Downloads/Spirochaete.fasta vagrant@127.0.0.1:/home/vagrant/
creating wrappers for the singularity commands
Here is an example bash script wrapper for a singularity execution of a command in a container.
new_assemblathon
#!/bin/bash
singularity exec ISUGIFsingularity-utilities-master.simg new_Assemblathon.pl
This wrapper contains the singularity command and once put in your path you can just use
new_Assemblathon
to execute the script via the singularity container.